
Principal InvestigatorInvestigador Principal
Jorge Doña
Associate Professor · University of GranadaProfesor Titular · Universidad de Granada
Ecology & evolution of bird symbiontsEcología y evolución de simbiontes de aves through molecular and genomic approachesmediante aproximaciones moleculares y genómicas

My research focuses on the ecology and evolution of bird symbionts using molecular and genomic approaches.
Mi línea de investigación se centra en la ecología y evolución de los simbiontes de las aves usando metodologías moleculares y genómicas.
I completed my PhD at the Doñana Biological Station (CSIC), where I studied the eco-evolutionary processes underlying host-shift speciation in highly host-specific symbionts, focusing on bird feather mites.
Realicé mi tesis doctoral en la Estación Biológica de Doñana (CSIC), donde investigué los procesos eco-evolutivos que subyacen a la especiación por cambio de hospedador en simbiontes altamente específicos (ácaros de las plumas de las aves).
I was a postdoctoral researcher at the University of Illinois at Urbana-Champaign under a Marie Skłodowska-Curie Global Fellowship, and I have been a Research Affiliate there since 2017.
Fui investigador postdoctoral en la Universidad de Illinois en Urbana-Champaign con una beca Marie Skłodowska-Curie Global Fellowship y soy Research Affiliate del centro desde 2017.
My postdoctoral research addressed fundamental yet poorly understood evolutionary processes in host-symbiont systems, particularly in avian lice, including the roles of microbiomes, hybridization, and genomic introgression.
Mi investigación postdoctoral abordó procesos evolutivos fundamentales pero poco estudiados en sistemas hospedador-simbionte, en particular en piojos de las aves, como el papel de los microbiomas, la hibridación y la introgresión genómica.
I am currently developing a new research line on climate-driven local genomic adaptation in obligate symbionts (GENCLIMLICE).
En la actualidad estoy desarrollando una nueva línea sobre la huella genómica de la adaptación local al clima en simbiontes permanentes (GENCLIMLICE).
I serve as Red List Coordinator for the IUCN SSC Parasite Specialist Group.
Soy Coordinador de la Lista Roja del IUCN SSC Parasite Specialist Group.
Climate-driven local genomic adaptation in highly host-specialized bird lice.Huella genómica de la adaptación local al clima en piojos de aves altamente especializados.
Conservation impacts of hybridization and introgression in symbionts: measuring the magnitude and role in shaping eco-evolutionary variables.Impacto de la hibridación y la introgresión en la conservación de organismos simbiontes: midiendo su magnitud y su papel en la variación eco-evolutiva.
Experimental evolution and functional genomics of lice — a project led by Dale H. Clayton and Sarah E. Bush (University of Utah). I collaborated on the comparative genomics of louse endosymbionts.Evolución experimental y genómica funcional de piojos — proyecto liderado por Dale H. Clayton y Sarah E. Bush (Universidad de Utah). Colaboré en la genómica comparada de endosimbiontes de piojos.
The lab studies how bird symbionts (feather mites and lice) evolve, adapt and diversify, combining fieldwork, molecular tools, genomics and bioinformatics. We welcome curious students interested in evolution, symbionts and genomics.El grupo estudia cómo evolucionan, adaptan y diversifican los simbiontes de las aves (ácaros de las plumas y piojos), combinando trabajo de campo, herramientas moleculares, genómica y análisis bioinformáticos. ¡Estudiantes con curiosidad por la evolución, los simbiontes y la genómica son bienvenidos!

At the University of Granada we are part of the Evolutionary Ecology & Behaviour (UGR) group.En la Universidad de Granada formamos parte del grupo Ecología Evolutiva y Comportamiento (UGR).
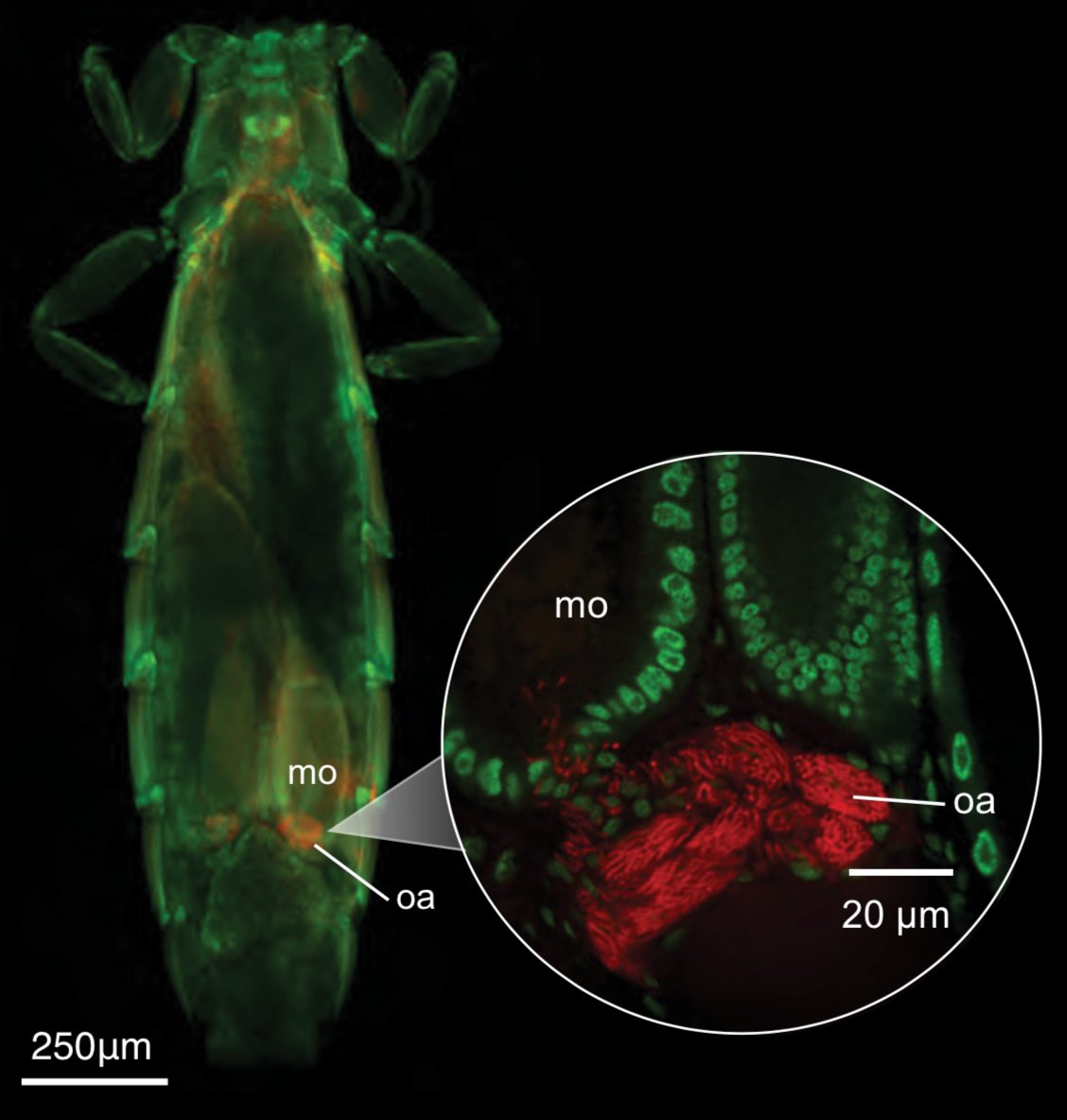
A riddle wrapped in an enigma: parasitic lice as clues to the evolutionary puzzle of Sapayoa (Aves).
Soto-Patiño, J., Doña, J., Johnson, K.P.
Biology Letters

Soto-Patiño, J., Walden, K.K.O., Doña, J., D'Alessio, L.M., Bush, S.E., Clayton, D.H., Dale, C., Johnson, K.P.
Royal Society Open Science

Doña, J., Jovani, R.
Ecosistemas
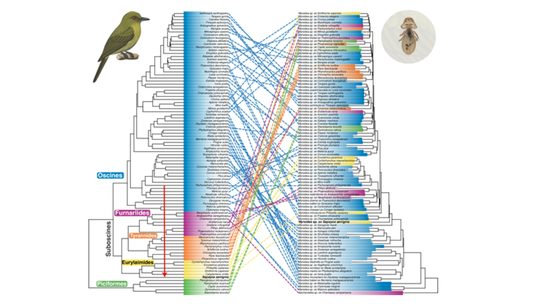
Clodfelter, E.B., Doña, J., Walden, K.K.O., Johnson, K.P.
Molecular Phylogenetics and Evolution
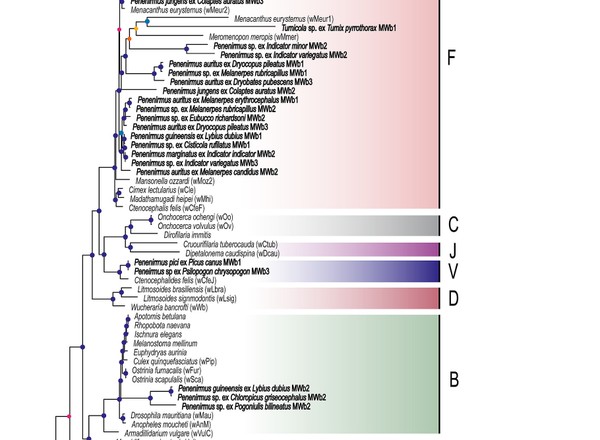
Phylogenomics reveals the timescale of diversification in Amblycera.
Najer, T., Doña, J., Buček, A., Sweet, A.D., Sychra, O., Johnson, K.P.
Systematic Entomology
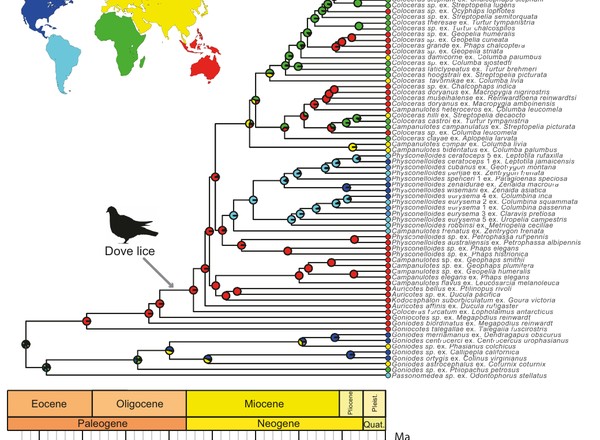
Sweet, A.D., Doña, J., Johnson, K.P.
Systematic Biology
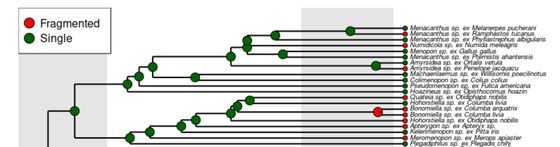
Mitochondrial genome fragmentation is correlated with increased rates of molecular evolution.
Najer, T., Doña, J., Buček, A., Sweet, A.D., Sychra, A., Johnson, K.P.
PLOS Genetics
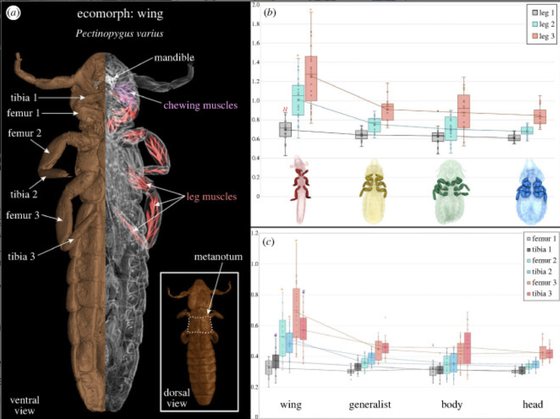
Parasite escape mechanisms drive morphological diversification in avian lice.
Kolencik, S., Stanley, E.L., Punnath, A., Grant, A.R., Doña, J., Johnson, K.P., Allen, J.M.
Proceedings B

Phylogenomics and biogeography of the feather lice (Phthiraptera: Ischnocera) of parrots.
Johnson, K.P., Doña, J.
Biological Journal of the Linnean Society

del Mar Labrador, M., Serrano, D., Doña, J., Aguilera, E., Arroyo, J.L., Atiénzar, F., … Jovani, R.
Journal of Animal Ecology

Doña, J., Johnson, K.P.
Evolution Letters
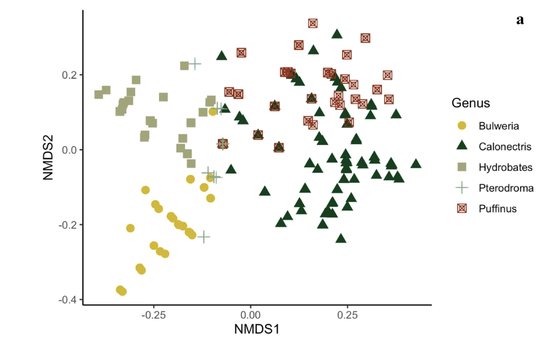
Stefan, L.M., Isbert, W., Gómez-Díaz, E., Mironov, S.V., Doña, J., McCoy, K.D., González-Solís, J.
Scientific Reports
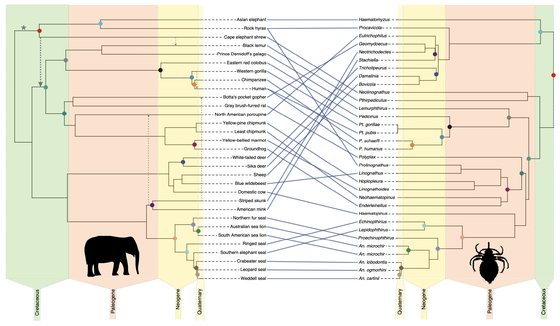
Phylogenomics reveals the origin of mammal lice out of Afrotheria.
Johnson, K.P., Matthee, C., Doña, J.
Nature Ecology & Evolution
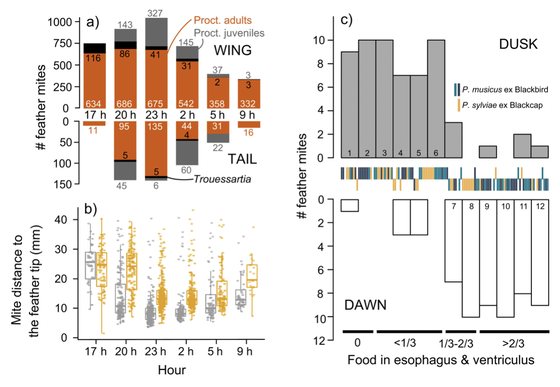
Feather mites at night: an exploration of their feeding, reproduction, and spatial ecology.
Labrador, M.M., Doña, J., Serrano, D., Jovani, R.
Ecology
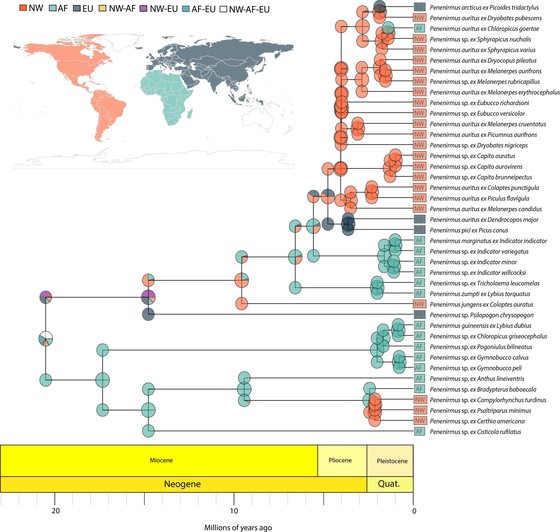
Johnson, K.P., Weckstein, J.D., Virrueta Herrera, S., Doña, J.
Molecular Phylogenetics and Evolution
Osuna-Mascaró, C., Doña, J., Johnson, K.P., De Rojas, M.
Microorganisms
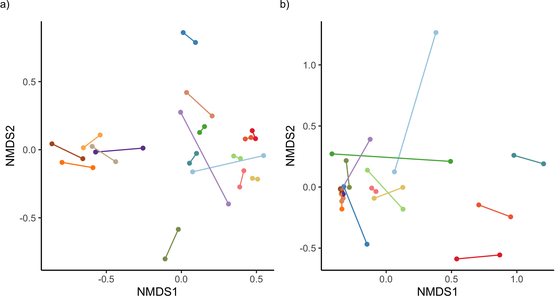
Patterns of microbiome variation among infrapopulations of permanent bloodsucking parasites.
Doña, J., Virrueta Herrera, S., Nyman, T., Kunnasranta, M., Johnson, K.P.
Frontiers in Microbiology
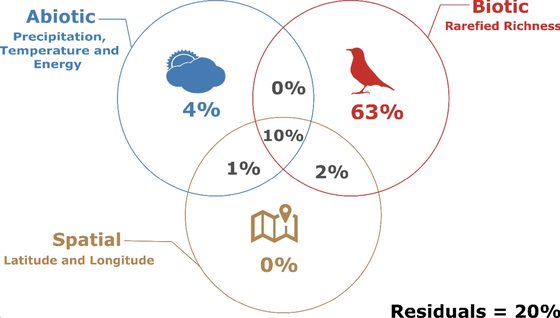
Gusmão, R.A.F., Hernandes, F.A., Vancine, M.H., Naka, L.N., Doña, J., Gonçalves-Souza, T.
Diversity and Distributions
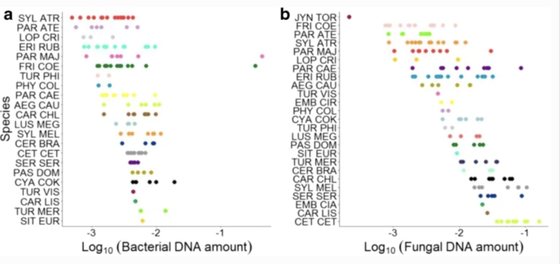
Labrador, M.M., Doña, J., Serrano, D., Jovani, R.
Microbial Ecology
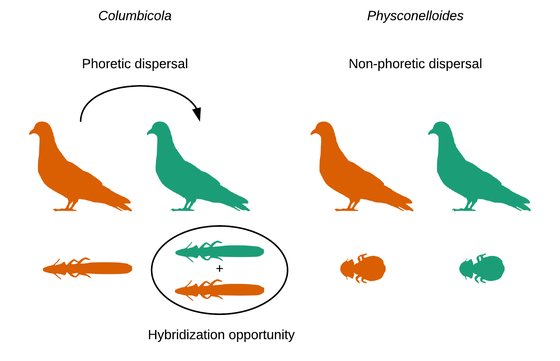
Comparing rates of introgression in parasitic feather lice with differing dispersal capabilities.
Doña, J., Sweet, A.D., Johnson, K.P.
Communications Biology

Contrasting stripes are a widespread feature of group living in birds, mammals and fishes.
Negro, J.J., Doña, J., Blázquez, M.C., Rodríguez, A., Herbert-Read, J.E., Brooke, M.d.L.
Proceedings B
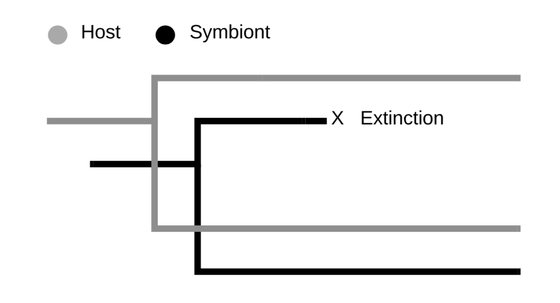
Assessing symbiont extinction risk using cophylogenetic data.
Doña, J., Johnson, K.P.
Biological Conservation
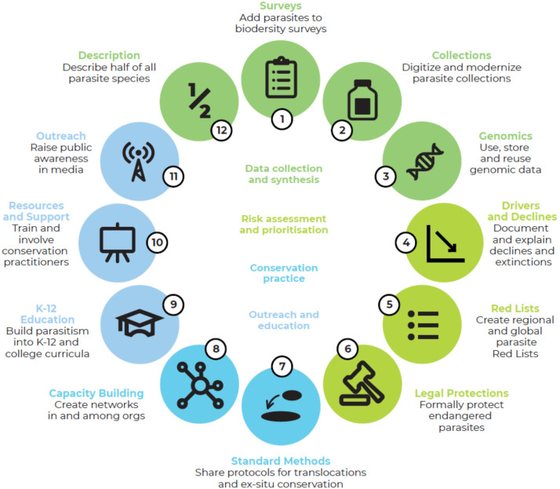
A global parasite conservation plan.
Carlson, C.J., Hopkins, S., Bell, K.C., Doña, J., Godfrey, S.S., Kwak, M., Lafferty, K.D., Moir, M.L., Speer, K.A., Strona, G., Torchin, M., Wood, C.
Biological Conservation
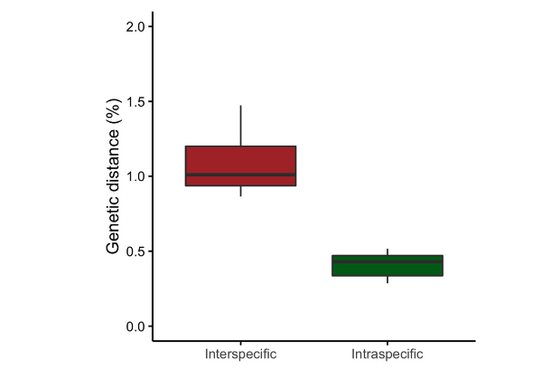
Márquez, F., Trovant, B., Van der Molen, S., Sepúlveda, R.D., Doña, J., Johnson, K.P., Vierna, J.
Organisms Diversity & Evolution
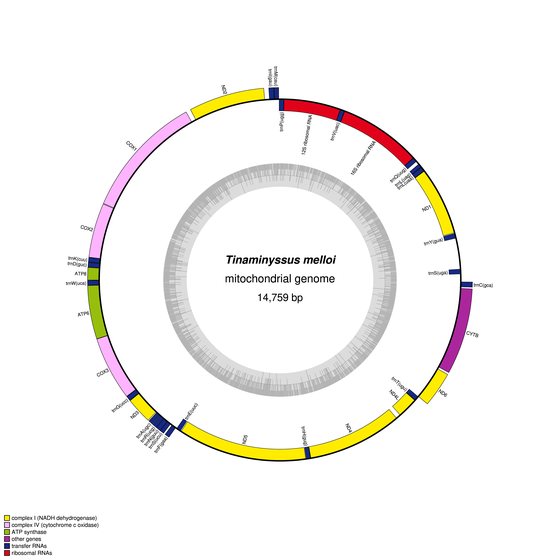
Osuna-Mascaró, C., Doña, J., Johnson, K.P., Esteban, R., De Rojas, M.
Frontiers in Ecology and Evolution

De Rojas, M., Doña, J., Dimov, I.
Biodiversity Data Journal
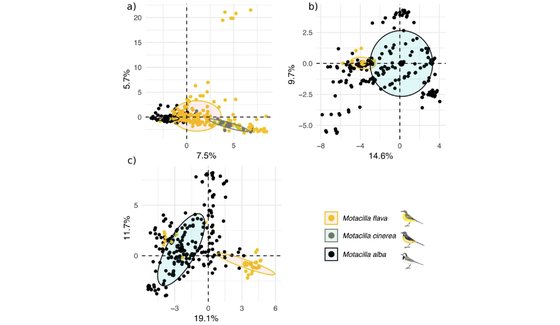
Persistence of single species of symbionts across multiple closely-related host species.
Doña, J., Osuna-Mascaró, C., Johnson, K.P., Serrano, D., Aymí, R., Jovani, R.
Scientific Reports

Doña, J., Serrano, D., Mironov, S., Montesinos-Navarro, A., Jovani, R.
Molecular Ecology
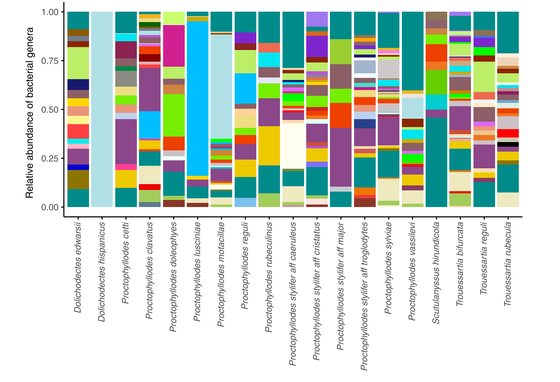
Doña, J., Proctor, H.C., Serrano, D., Johnson, K.P., Oddy-van Oploo, A., Ascunce, M.S., Huguet-Tapia, J.C., Jovani, R.
Molecular Ecology
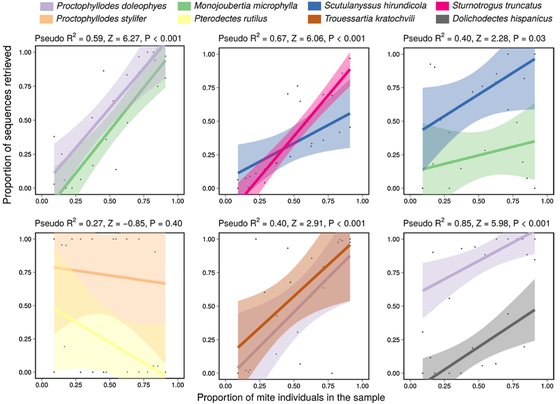
Enabling large-scale feather mite studies: an Illumina DNA metabarcoding pipeline.
Vizcaíno, A., Doña, J., Vierna, J., Marí-Mena, N., Esteban, R., Mironov, S., Urien, C., Serrano, D., Jovani, R.
Experimental and Applied Acarology
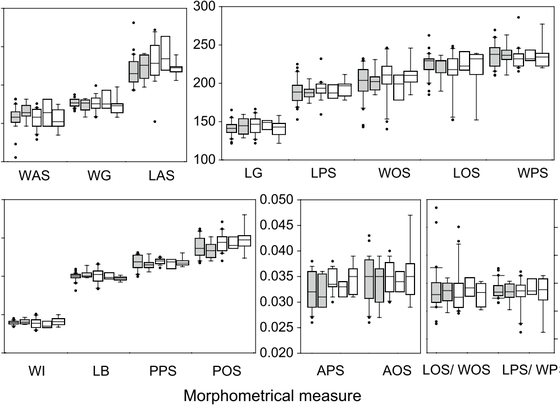
De Rojas, M., Doña, J., Jovani, R., Dimov, I., Zurita, A., Callejón, R., Rodríguez-Plá, M.
Experimental and Applied Acarology
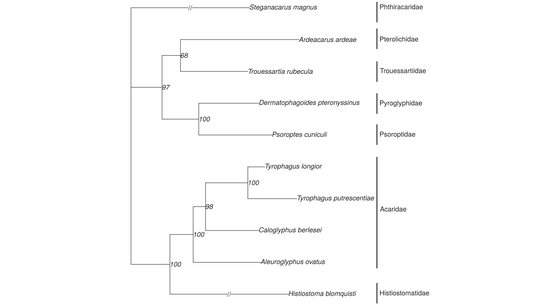
Esteban, R., Doña, J., Vierna, J., Vizcaíno, A., Serrano, D., Jovani, R.
Mitochondrial DNA Part B
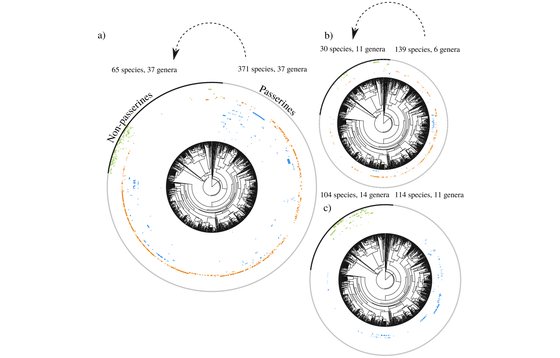
Doña, J., Proctor, H.C., Mironov, S.V., Serrano, D., Jovani, R.
Global Ecology and Biogeography
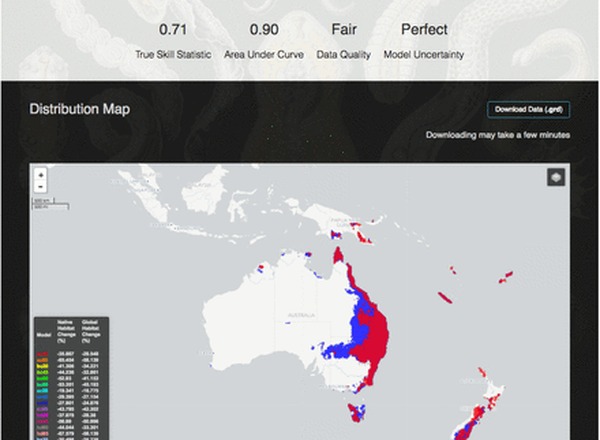
Carlson, C.J., Muellerklein, O.C., Phillips, A.J., Burgio, K.R., Castaldo, G., Cizauskas, C.A., Cumming, G.S., Dallas, T.A., Doña, J., Harris, N., Jovani, R., Miao, Z., Proctor, H., Yoon, H.S., Getz, W.M.
BioRxiv
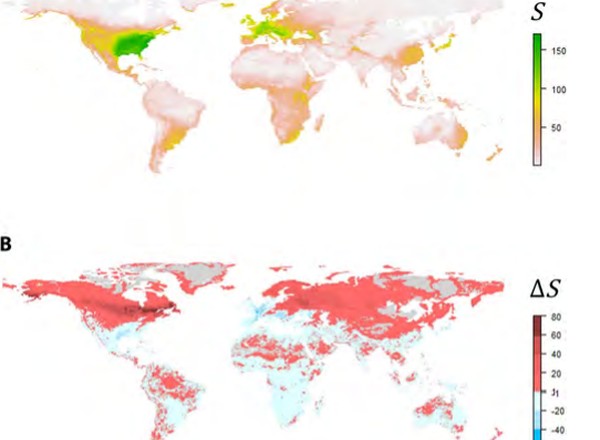
Parasite biodiversity faces extinction and redistribution in a changing climate.
Carlson, C., Burgio, K., Dougherty, E., Phillips, A., Bueno, V., Clements, C., Castaldo, C., Dallas, T., Cizauskas, C., Cumming, C., Doña, J., Harris, N., Jovani, R., Mironov, S., Muellerklein, O., Proctor, H., Getz, W.
Science Advances
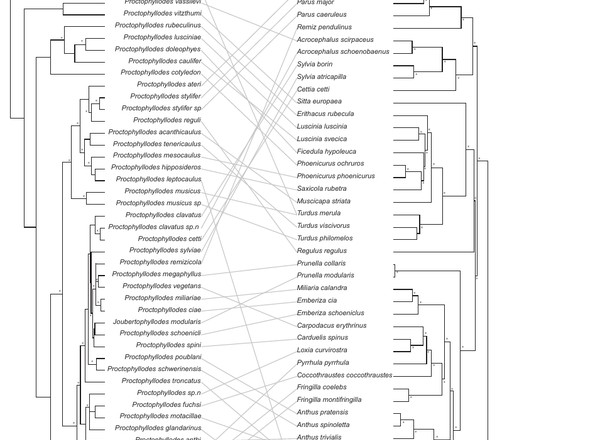
Doña, J., Sweet, A.D., Johnson, K.P., Serrano, D., Mironov, S.V., Jovani, R.
Molecular Phylogenetics and Evolution
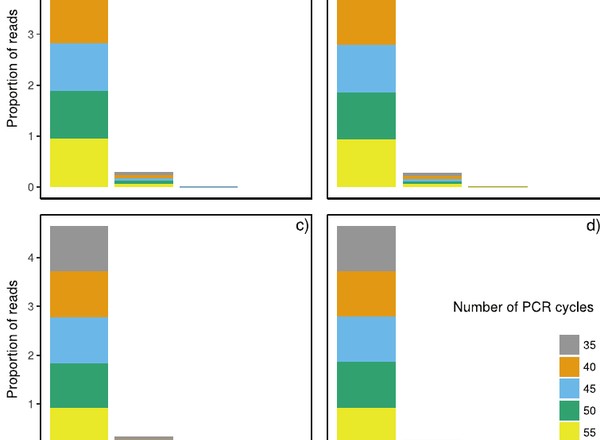
PCR cycles above routine numbers do not compromise high-throughput DNA barcoding results.
Vierna, J., Doña, J., Vizcaíno, A., Serrano, D., Jovani, R.
Genome

Opening the doors of parasitology journals to other symbionts.
Jovani, R., Doña, J., Labrador, M., Serrano, D.
Trends in Parasitology
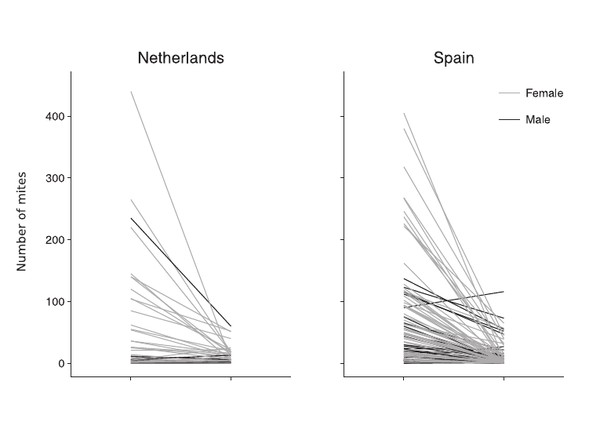
Vertical transmission in feather mites: insights into its adaptive value.
Doña, J., Potti, J., De la Hera, I., Blanco, G., Frias, O., Jovani, R.
Ecological Entomology
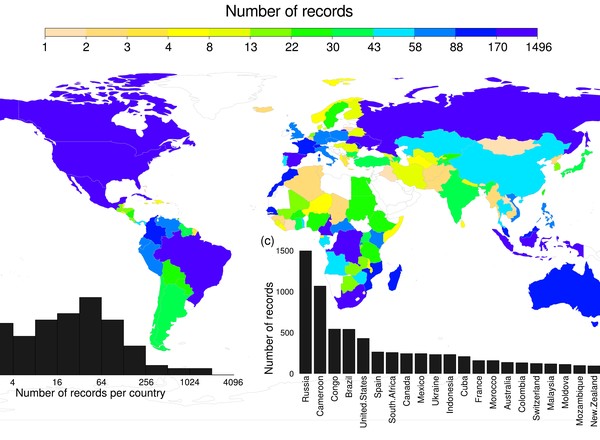
Global associations between birds and vane-dwelling feather mites.
Doña, J., Proctor, H., Mironov, S., Serrano, D., Jovani, R.
Ecology
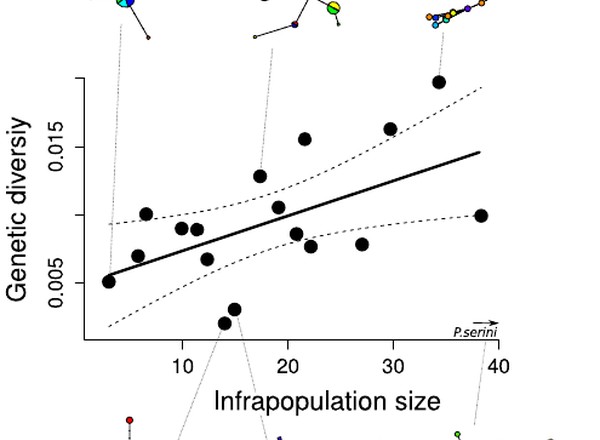
Species mtDNA genetic diversity explained by infrapopulation size in a host-symbiont system.
Doña, J., Moreno-García, M., Criscione, C.D., Serrano, D., Jovani, R.
Ecology and Evolution
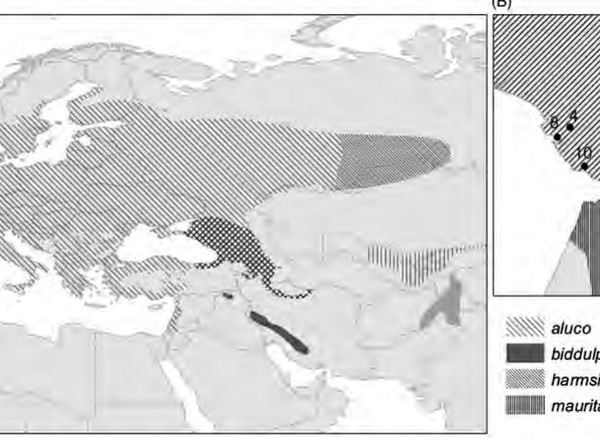
Doña, J., Ruiz-Ruano, F.J., Jovani, R.
Mitochondrial DNA Part A
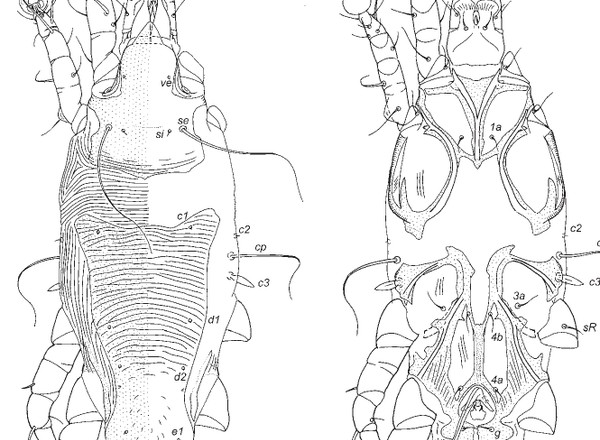
Mironov, S., Doña, J., Jovani, R.
Folia Parasitologica
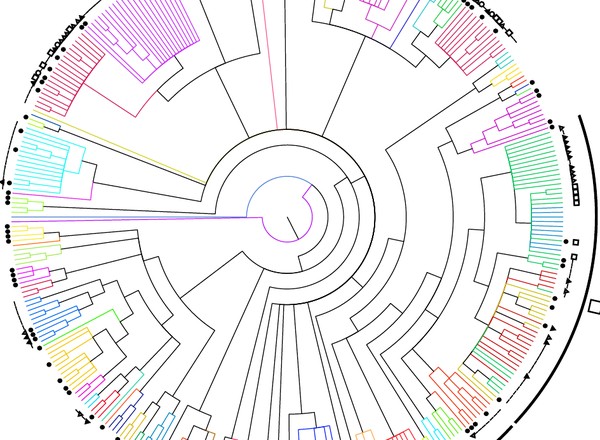
DNA barcoding and mini-barcoding as a powerful tool for feather mite studies.
Doña, J., Diaz-Real, J., Mironov, S., Bazaga, P., Serrano, D., Jovani, R.
Molecular Ecology Resources
Related to my research, I also enjoy nature photography, mostly birds.Relacionado con mi investigación, también me gusta la fotografía de naturaleza, sobre todo de aves.

















Collaborations, prospective students, or anything about my research, reach out.Colaboraciones, estudiantes interesados o cualquier cosa sobre mi investigación, escríbeme.
jorged@ugr.es