Enabling large-scale feather mite studies: An Illumina DNA metabarcoding pipeline
Enabling large-scale feather mite studies: An Illumina DNA metabarcoding pipeline
Abstract
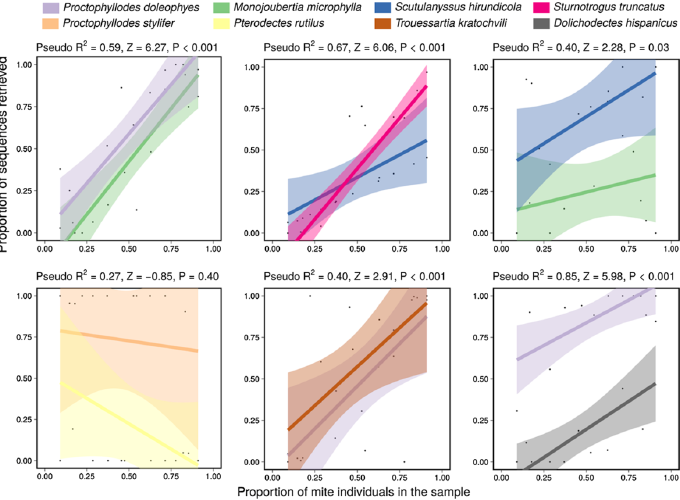
Feather mites are among the most common and diverse ectosymbionts of birds, yet basic questions such as the nature of their relationship remain largely unanswered. One reason for feather mites being understudied is that their morphological identification is often virtually impossible when using female or young individuals. Even for adult male specimens this task is tedious and requires advanced taxonomic expertise, thus hampering large-scale studies. In addition, molecular-based methods are challenging because the low DNA amounts usually obtained from these tiny mites do not reach the levels required for high-throughput sequencing. This work aims to overcome these issues by using a DNA metabarcoding approach to accurately identify and quantify the feather mite species present in a sample. DNA metabarcoding is a widely used molecular technique that takes advantage of high-throughput sequencing methodologies to assign the taxonomic identity to all the organisms present in a complex sample (i.e., a sample made up of multiple specimens that are hard or impossible to individualise). We present a high-throughput method for feather mite identification using a fragment of the COI gene as marker and Illumina Miseq technology. We tested this method by performing two experiments plus a field test over a total of 11,861 individual mites (5360 of which were also morphologically identified). In the first experiment, we tested the probability of detecting a single feather mite in a heterogeneous pool of non-conspecific individuals. In the second experiment, we made 2×2 combinations of species and studied the relationship between the proportion of individuals of a given species in a sample and the proportion of sequences retrieved to test whether DNA metabarcoding can reliably quantify the relative abundance of mites in a sample. Here we also tested the efficacy of degenerate primers (i.e., a mixture of similar primers that differ in one or several bases that are designed to increase the chance of annealing) and investigated the relationship between the number of mismatches and PCR success. Finally, we applied our DNA metabarcoding pipeline to a total of 6501 unidentified and unsorted feather mite individuals sampled from 380 European passerine birds belonging to 10 bird species (field test). Our results show that this proposed pipeline is suitable for correct identification and quantitative estimation of the relative abundance of feather mite species in complex samples, especially when dealing with a moderate number (>30) of individuals per sample.